5. Visium Breast cancer¶
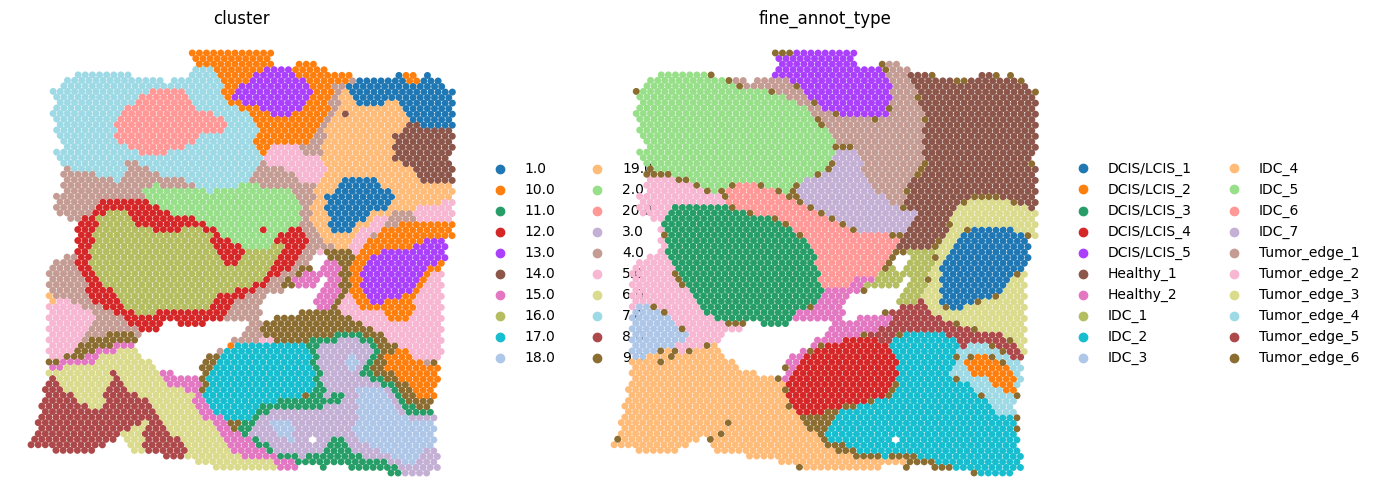
We applied SECE to breast cancer data sequenced by Visium. Experts have annotated the results according to the H&E staining and obtained pathology annotation containing 4 phenotypes and 20 subdivisions, they were ductal carcinoma in situ/lobular carcinoma in situ (DCIS/LCIS), healthy tissue (Healthy), invasive ductal carcinoma (IDC), and tumor surrounding regions with low features of malignancy (Tumor edge). Raw data was download from https://www.10xgenomics.com/resources/datasets/human-breast-cancer-block-a-section-1-1-standard-1-1-0.
Load data¶
[1]:
import os
import warnings
warnings.filterwarnings('ignore')
import SECE
import torch
import numpy as np
import scanpy as sc
import random
result_path = 'breast_cancer'
os.makedirs(result_path, exist_ok=True)
[2]:
adata = sc.read('./data/breast_cancer.h5ad')
Creating and training the model¶
[3]:
sece = SECE.SECE_model(adata.copy(),
result_path=result_path,
hvg = 2000,
dropout_rate=0.1,
dropout_gat_rate = 0.2,
device='cuda:0')
Likelihood: nb
Input dim: 2000
Latent Dir: 32
Model1 dropout: 0.1
Model2 dropout: 0.2
AE Module of SECE¶
[4]:
sece.prepare_data(lib_size='explog')
Library size: explog
Input normalize: True
Input scale: False
Hvg: 2000
(3798, 2000)
[5]:
sece.train_model1(epoch1=50, plot=True)
100%|████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 50/50 [00:28<00:00, 1.75it/s]
Model1 lr: 0.001
Model1 epoch: 50
Model1 batch_size: 128
[6]:
adata1 = sece.predict_model1(batch_size=128)
GAT Module of SECE¶
[7]:
sece.prepare_graph(cord_keys=['x','y'],
latent_key = 'X_CE',
num_batch_x=1,
num_batch_y=1,
neigh_cal='knn',
n_neigh=6,
kernal_thresh=1)
Batch 1: Each cell have 6.0 neighbors
Batch 1: Each cell have 6similar cells
All: Each cell have 6.0 neighbors
Graph cal: knn
knn: 6
kernal_thresh: 1
[8]:
sece.train_model2( lr_gat=0.01,
epoch2=40,
re_weight=1,
si_weight=0.3,
plot=True)
100%|████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 40/40 [00:02<00:00, 19.54it/s]
Model2 lr: 0.01
Model2 epoch: 40
Model2 similar weight: 0.1
[9]:
adata1 = sece.predict_model2()
Spatial domains¶
Clustering¶
[10]:
SECE.cluster_func(adata1, clustering='mclust', use_rep='X_SE', cluster_num=20, key_add='cluster')
VSCode R Session Watcher requires jsonlite, rlang. Please install manually in order to use VSCode-R.
R[write to console]: __ __
____ ___ _____/ /_ _______/ /_
/ __ `__ \/ ___/ / / / / ___/ __/
/ / / / / / /__/ / /_/ (__ ) /_
/_/ /_/ /_/\___/_/\__,_/____/\__/ version 5.4.10
Type 'citation("mclust")' for citing this R package in publications.
fitting ...
|======================================================================| 100%
[10]:
AnnData object with n_obs × n_vars = 3798 × 2000
obs: 'in_tissue', 'array_row', 'array_col', 'annot_type', 'fine_annot_type', 'x', 'y', 'size', 'n_counts', 'cluster'
var: 'gene_ids', 'feature_types', 'genome', 'n_cells', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm'
uns: 'spatial', 'log1p', 'hvg'
obsm: 'spatial', 'spatial1', 'X_CE', 'X_SE'
layers: 'counts', 'expr'
Visualization¶
[11]:
import matplotlib.pyplot as plt
from matplotlib.pyplot import rc_context
with rc_context({'figure.figsize': (6,6)}):
sc.pl.embedding(adata1, basis='spatial1', color=['cluster','fine_annot_type'],
frameon=False, size=100, show=True)
[13]:
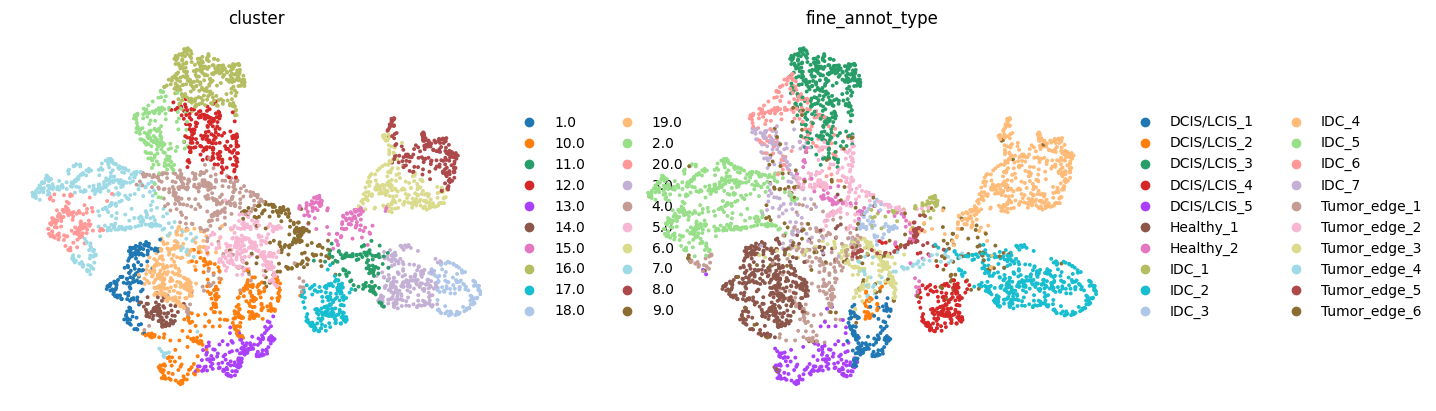
sc.pp.neighbors(adata1, use_rep='X_SE', key_added='X_SE')
adata1.obsm['umap_SE'] = sc.tl.umap(adata1, neighbors_key='X_SE', copy=True).obsm['X_umap']
sc.pl.embedding(adata1, basis='umap_SE', color=['cluster','fine_annot_type'],
frameon=False, ncols=2, show=False)
[13]:
[<AxesSubplot: title={'center': 'cluster'}, xlabel='umap_SE1', ylabel='umap_SE2'>,
<AxesSubplot: title={'center': 'fine_annot_type'}, xlabel='umap_SE1', ylabel='umap_SE2'>]
[14]:
adata1.write(f'{result_path}/adata1.h5ad')